
DNA methylation refers to addition of a methyl group at the carbon-5 site of cytosine residues of the CpG dinucleotide by DNA methyltransferases (DNMTs). CpG dinucleotide accounts for ~1% of human genome and 60%-90% of all CpGs are methylated. Unmethylated CpGs are grouped in clusters called "CpG islands" that are present in the 5' regulatory regions of many genes. DNA methylation can repress gene expression by blocking the binding of transcription factors or modifying chromatin structure to a repressive state.
It has become increasingly evident that alteration in DNA methylatioin has a profound role in the pathogenesis of human diseases, such as cancer and neurodegenerative disorders. The role of DNA methylation in cancer has been relatively well studied. While wide spread DNA hypomethylation can lead to chromosomal instability and inappropriate gene activation, tumor suppressor genes can show promoter hypermethylation and gene inactivation, which contribute to tumour development.
By Barros & Offenbacher J Dent Res 2009; 88: 400
Several studies have suggested that DNA methylation plays a significant role in osteogenic cell differentiation and bone metabolism. For example, Kitazawa et al. (Mol.Endocrinol., 2007. 21: 148-158) analyzed the effect of DNA methylation on the cell- and tissue-specific expression of RANKL gene and osteoclastogenesis. Higher level of CpG methylation at the RNAKL promoters was detected in various tissues expressing no/lower level of RANKL, and in subpopulations of stromal/osteoblastic cells that barely supports osteoclastogenesis. In vitro methylation of the RANKL gene promoter construct in stromal/osteoblastic cells resulted in reduced transcriptional activity and poor response to vitamin D3. In contrast, treatment with DNA methyltransferase inhibitor significantly restored RANKL expression and osteoclastogenesis. These results suggested that CpG methylation of the RANKL gene promoter reversibly suppresses RANKL gene expression, and the heterogeneity of stromal/osteoblastic cells in response to bone-resorbing stimuli may be attributed, in part, to the methylation status of the RANKL gene promoter. Additionally, previous studies have demonstrated that CpG methylation at promoters of the osteocalcin (OC) gene (Villagra A, et al., J Cell Biochem., 2002. 85: 112-122) and the estrogen receptor alpha (ESR1) gene (Penolazzi L., et al., J Steroid Biochem Mol Biol., 2004. 91: 1-9) may influence their gene expression in human osteoblastic cells and osteoblast differentiation.
Therefore, it is imperative to ascertain genomic DNA methylation patterns in osteogenic cells, identify their targeted genes, and determine their relationship to BMD variation. This project holds a great promise of award to yield ground breaking outcomes in the osteoporosis research field. The results will lead to identification of novel heritable factors contributing to human BMD variation and osteoporosis risks. The study may thus lead to a major paradigm shift to greatly expand human genetic studies of osteoporosis from classical DNA sequence variants to novel epigenetics/epigenomics mechanisms of heritable risk to osteoporosis in vivo in humans.
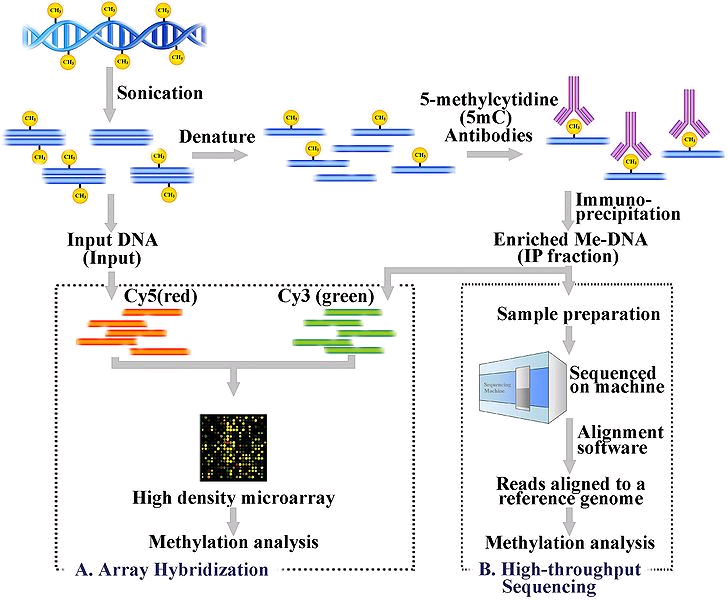
Workflow of MeDIP-chip /-Seq.
Despite these pioneering DNA methylation studies in the bone field, genome-wide DNA methylation patterns – the methylome – in primary human osteogenic cells is poorly understood. More importantly, the role of DNA methylation in the pathophysiology of osteoporosis in humans is largely unknown at this time. To address these questions, we recently carried out a pilot comparative methylome profiling analysis in peripheral blood monocytes (PBMs, potential precursors of bone-resorbing osteoclasts), using the MeDIP-Seq method (methylated DNA immunoprecipitation coupled with the next-generation sequencing).
Differential methylated regions (DMRs) related to BMD variation will be identified by comparing methylome pattern in PBMs from subjects with low BMD vs. high BMD. Furthermore, by integrating the methylome data with transcriptome data and conducting molecular functional studies, we will identify the DMR targeted genes and determine how these genes contribute to human BMD variation and osteoporosis risks.
